Gene Fusions Tutorial - DISCASM and GMAP-Fusion
DISCASM and GMAP-Fusion Tutorial
DISCASM and GMAP-fusion are software focused on exploring transcripts that are ill-represented within a reference genome, either lacking genome representation (missing genes, foreign transcripts) or otherwise not well represented in the reference genome sequence due to chromosomal rearrangements. This is highly relevant for cancer biology for detecting fusion transcripts and oncoviruses from RNA-Seq data. DISCASM and GMAP-Fusion were developed as components of the broader Trinity Cancer Transcriptome Analysis Toolkit (CTAT). Each tool is briefly described below.
-
DISCASM targets RNA-Seq reads that are discordantly aligned to the genome or lacking alignment to the genome altogether for de novo transcriptome assembly. De novo transcriptome assembly is performed using either Trinity or Oases.
-
GMAP-fusion use the GMAP aligner to identify candidate fusion transcripts followed by additional analysis to weed out likely artifacts and further score fusion candidates based on RNA-Seq read supporting evidence.
These tools can be used separately or synergistically. In this tutorial, we showcase the latter, where we use DISCASM to first assemble discordant and unmapped reads, and then use GMAP-fusion to identify reconstructed fusion transcripts. We also further explore the reconstructed transcripts from discordant and genome-unmapped reads to see if we have evidence of foreign transcripts such as derived from cancer-relevant viruses. For exploring foreign transcripts, we leverage Centrifuge.
Tutorial Software and Data Setup
All software and data required to run through the tutorial is installed locally on the server. You just need to copy it over to your workspace like so:
% cp -r ~/CourseData/CG_data/Module9/GMAP-fusion .
This will add the tools GMAP-fusion and DISCASM along with the required tutorial data.
We next just need to do a little environment configuration so that the system will find the required software.
Set up the following environmental variables like so:
% cd GMAP-fusion/
% export GMAP_FUSION_HOME=`pwd`
% export DISCASM_HOME=`pwd`/DISCASM
Tutorial data contents
Next enter the Tutorial/ subdirectory
% cd Tutorial
The data here correspond to a small region of human chromosome 2 and RNA-Seq reads derived from a breast cancer cell line HCC1395, and further supplemented with data from another cell line SKBR3 provided by Henrik Edgren et al.
Note, parts of this tutorial are based on earlier work by Andrew McPherson using cell line data generated by Malachi Griffith.
List the files in the Tutorial directoryL
% ls -l
-rw-rw-r-- 1 ubuntu ubuntu 2488924 Mar 14 10:14 HCC1395-miniplus_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 74 Mar 14 10:14 README.md
-rw-rw-r-- 1 ubuntu ubuntu 2500464 Mar 14 10:14 HCC1395-miniplus_2.fastq.gz
-rwxrwxr-x 1 ubuntu ubuntu 195 Mar 14 10:14 cleanMe.sh
-rw-rw-r-- 1 ubuntu ubuntu 1565706 Mar 14 10:14 minichr2.fa
-rw-rw-r-- 1 ubuntu ubuntu 684002 Mar 14 10:14 minichr2.gtf
-rw-rw-r-- 1 ubuntu ubuntu 98099026 Mar 14 10:14 centrifuge_VirusDB.tar.gz
drwxrwxr-x 2 ubuntu ubuntu 4096 Mar 14 10:14 centrifuge_VirusDB
Data provided in the tutorial specifically include:
minichr2.fa : short segment of human chr2
minichr2.gtf : corresponding gene annotations for this region of chr2
HCC1395-miniplus_1.fastq.gz : 'left' fragment cancer line RNA-Seq reads
HCC1395-miniplus_2.fastq.gz : 'right' fragment cancer line RNA-Seq reads
Steps of the Tutorial
The following tutorial will take you through preparing the above provided genome and annotation files for use with Trinity CTAT tools, followed by aligning reads, capturing the discordant and unmapped reads, performing de novo transcriptome assembly, and identifying reconstructed fusion transcripts as well as any interesting transcripts supported by the RNA-Seq data but not represented by the target genome.
Prep the genome for use with Trinity CTAT
Run the following to prepare the genome and annotations for use with GMAP-Fusion, DISCASM, STAR, and other processes we’ll need for fusion discovery and analysis as part of Trinity CTAT.
% $GMAP_FUSION_HOME/FusionFilter/prep_genome_lib.pl \
--genome_fa minichr2.fa \
--gtf minichr2.gtf \
--gmap_build
Align RNA-Seq reads to the genome
Use STAR to align the RNA-Seq data to the genome:
STAR --genomeDir ctat_genome_lib_build_dir/ref_genome.fa.star.idx \
--readFilesIn HCC1395-miniplus_1.fastq.gz HCC1395-miniplus_2.fastq.gz \
--outReadsUnmapped None \
--chimSegmentMin 12 \
--chimJunctionOverhangMin 12 \
--alignSJDBoverhangMin 10 \
--alignMatesGapMax 100000 \
--alignIntronMax 100000 \
--alignSJstitchMismatchNmax 5 -1 5 5 \
--runThreadN 4 \
--outSAMstrandField intronMotif \
--outSAMunmapped Within \
--outSAMtype BAM Unsorted \
--outSAMattrRGline ID:GRPundef \
--chimSegmentReadGapMax 3 \
--genomeLoad NoSharedMemory \
--twopassMode Basic \
--readFilesCommand 'gunzip -c'
# takes less than a minute
There are two important output files that will be generated by the above alignment command:
Aligned.out.bam : bam file containing the aligned reads
Chimeric.out.junction : identification of reads aligning discordantly to the genome
We’ll use both of these files when running DISCASM below.
De novo assemble discordant and genome-unmapped reads using DISCASM
Assemble the discordantly-aligned and genome-unmapped reads using DISCASM with the Trinity assembler, leveraging the outputs from STAR above like so:
% $DISCASM_HOME/DISCASM --chimeric_junctions Chimeric.out.junction \
--aligned_bam Aligned.out.bam \
--denovo_assembler Trinity \
--left_fq HCC1395-miniplus_1.fastq.gz \
--right_fq HCC1395-miniplus_2.fastq.gz \
--out_dir discasm
# takes a few minutes
Once the above completes, you should find the de novo assembled transcripts at:
discasm/trinity_out_dir/Trinity.fasta
Take a look at the file:
% head discasm/trinity_out_dir/Trinity.fasta .
>TRINITY_DN0_c0_g2_i1 len=243 path=[0:0-242]
GCCCAGGAGGCAGAGGCTTCAGTGAGCTGAGATTGTGCTACTATACTCCAACCTGGGTGAGTGTGAGAATACAGGTGTGCACACCACACTGCACTGCTTTTTAAATTTTTTGTAGATGTGAAGTCTCACTGTATTGACCAGGCTGGTCTCGAACTTAGGAGATCAAGCAGTCTTCCTGCCTCACCCTCCAAAGGTGCTGGGATTACAGGCATGAGCTACTGCACCTGGCCAGAGGCAACATTA
>TRINITY_DN11_c0_g1_i1 len=231 path=[1:0-230]
CTTGGTGGATTAACACAGCAAAGGTTTATTTCTCACCCACACCTGCACGGGCTGGCAAGAGGACTTTGCTCTGTTCAGTCATTCATGGGTCCATGCTTCTCCTATATAGCCAGCCCTCTGTCTCCTAAGGCCTGGAGTCCTCCATGGGCCTCCAATCTGTGGCTGACAGAGGATGTTGACTTGAGACCCATCTTTGATTCATTATGGAGCTTCCACAGGCTGTTGAAATCC
>TRINITY_DN11_c0_g1_i2 len=187 path=[0:0-186]
CTTTGTGTTTGAGAGGTTGGGAAAGAAGAACCAGGGGTGCTCTCAGGTAAATCAGTTTTAAGAAAAAATATACATGAAGATTCTGGAAATTAAGTTTCTTAAATACCCATGCTCCTGTATTCCTTAGGCATGTGTCCCCAGTTTGAGGACTTTTGACTTAGAGGATTTCAACAGCCTGTGGAAGCTC
>TRINITY_DN11_c0_g2_i1 len=261 path=[0:0-260]
TTGATTTGGCCTCATTCTGGCTGTCCCTGGTGCTGTCTTATCTCTTGAGAACTGGCTGTGTCTCCTGTTGAATTCCCTATCCGAACACTGCAGTTCTTGGCTACAAGGCAGTCACTTCACCCCTGCAGTGGTGAGATCCCATTAGCTACCATTAGCTACTCTTTTCGTCCAGAAAGGCGAAATGCCATAGCAAACAATCCCACAGTCTTGGTGGATTAACACAGCAAAGTCCTCTTGCCAGCCCATGCAGGTGTGGGTGAG
>TRINITY_DN12_c1_g3_i1 len=113 path=[0:0-112]
AGTATGATGTATGCTTTGTGCTGTTCTTCCAGCAATATCTGTAGTATGATAAGCTATTTGTCTCCTTGTTTCTCTAAATAAATGTTCCCATACAGCTCTTCCTACTATATTAT
Mine the reconstructed transcripts for fusions using GMAP-fusion
Discordantly-aligned reads are a signature for fusion transcripts. See if we were able to assembly an fusion transcripts from the discordantly (or unmapped) reads by running GMAP-fusion on the reconstructed transcripts:
% $GMAP_FUSION_HOME/GMAP-fusion \
-T discasm/trinity_out_dir/Trinity.fasta \
--left_fq HCC1395-miniplus_1.fastq.gz \
--right_fq HCC1395-miniplus_2.fastq.gz \
--genome_lib_dir ctat_genome_lib_build_dir/ \
--output gmapf
# takes a couple seconds
The fusion predictions will be found as file ‘gmapf/GMAP-fusion.final’.
Examine the fusion predictions. Do you find any?
% cat gmapf/GMAP-fusion.final
.
#fusion_name J S trans_acc trans_brkpt geneA chrA coordA geneB chrB coordB junction_type
PLA2R1--RBMS1 199 41 TRINITY_something... 272-271 PLA2R1 chr2 272580 RBMS1 chr2 599999 ONLY_REF_SPLICE
Exploring fusions using the UCSC Genome Browser
From the ‘discasm/trinity_out_dir/Trinity.fasta’ file, locate the fasta-formatted sequence for the transcript predicted as a fusion. (Try using ‘less’ unix utility and searching for the entry).
if you can’t easily locate yours, here’s mine
Visit the UCSC Genome Browser.
From the menu, go to ‘Tools’->’Blat’
Paste your fusion transcript into the form field and submit.
You should find the following results similar to this:
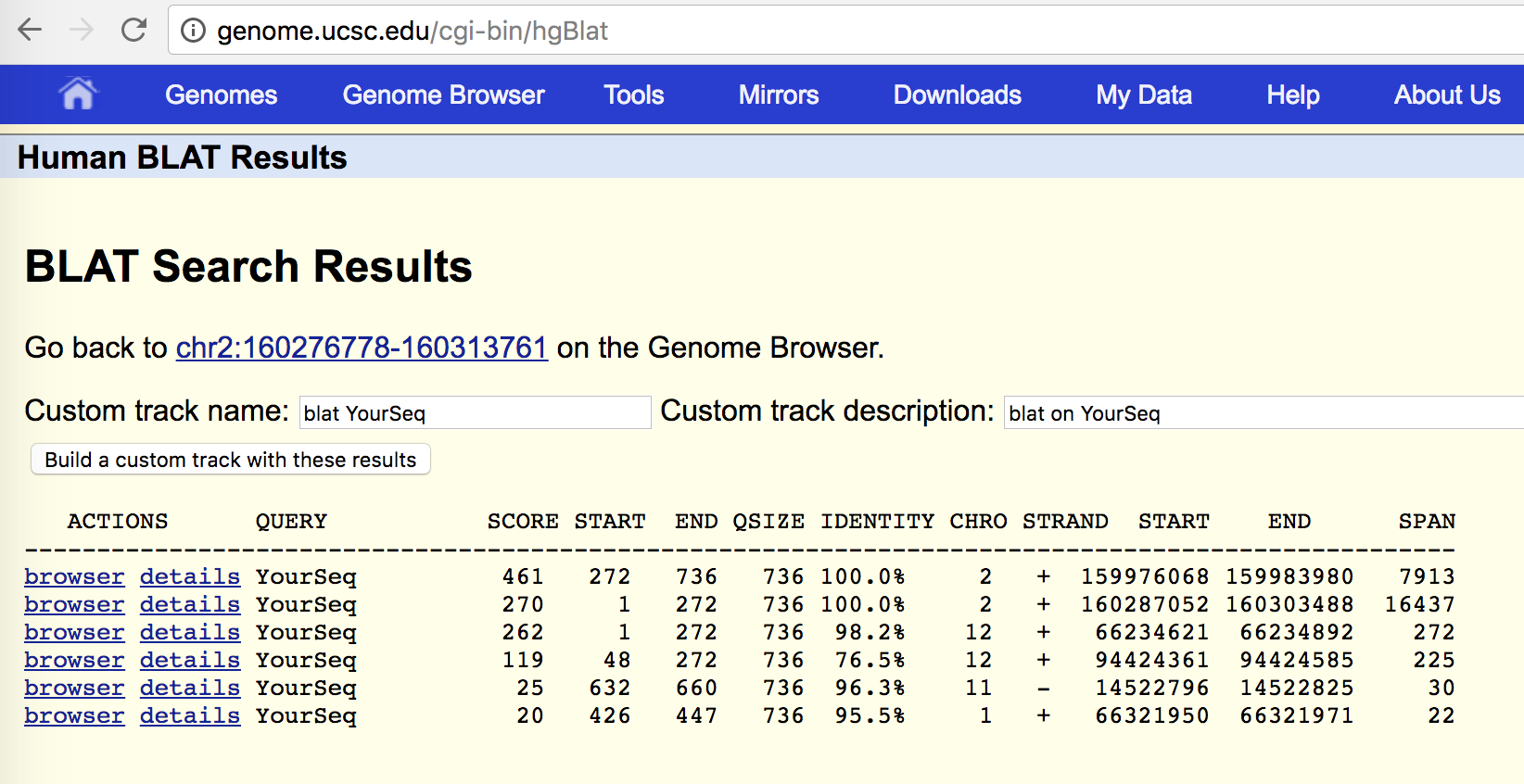
Explore the top two hits that are 100% identical to regions of chromosome 2.
Which genes do they match? Can you infer where the fusion breakpoint is based on the transcript alignments?
Comparison of fusion transcript with DNA-based fusion breakpoint
Earlier work by others using WGS sequence data and the destruct software yielded the following sequence supporting the physical breakpoint in the genome.
>destruct_31240
TGCAAAAGATCTGGAAAAATGCAGTCTGGTATTTACACATAATTTAAGTTCACAGTGC
AACTGCTCCCATAACCCTAGCTGAAACTGTCTCTTCTTAGTCATTTTTAATTTTCCAA
GATAACTTGGCAAAGCTATTGTTGTTGACATAATAAAGACTGGGCAGAAGGCTTACCT
AGCAAAGCCAACACCACGACTTGTACCACTGGAATCACGTAGTATCCTTGTAGAAATA
ACTTGTCCAAATGGTTTGAGCATATTTTCTAGTTCTTGCTCATCCATGGAGAGTGGCA
AATTAGAAATGTAGAGGTTGGTAGGATCTTGTTCCTGTTGCTAAAACAGAAGAGAGTG
TTGTCCATTAATTTCCAACAGAAGGTGAGATATTTATGTTAACACACCTATTTTTATT
AGCTACTTTCTTTGCTCAAGTCCTTTTAAAGTACTCAGAACCTCAGAACACCAAAGTC
ACCCTGGACTCTTGAAAATAGTGTCTGAAGCTTGGACAA[AA]AAAAAGTAATATTAG
AAAATGAATTCATTTTCTGACAAAAAATTATTGGCTCATCCTCTCAGTTATTTACCCT
CTCAGTGATTTATAATTCATTGCATATGTCACATGTATTTGAAAAACAATTCAAGGTA
TCAAGGCATCATTAGTATAAAGATACTGATTTTAGGTATTAGTCTGATTGCTAAGCTT
TAAGCAGTATAAGCTTTCCTTCCCATTCAAATAGAGAGACACAATATAGGACAAAAGA
ATACTACAGAGTGCCCAGTGTTTGACAACTAGAAAATTATCCTTTTGATGAGTTCATG
TCCTTTGCAGGGACATGGATGAAGCTGGAAACCATCAATCTCAGCAAACTAACACATG
AACAGAAAACCAAACACCGCATGTTCTCACTCATAAGTGGGAGCTGAACAATGAGAAC
ACATGGACACAGGGAGGGGAACATCACACACTGGGGCCTGTCGG
Use the UCSC Blat utility to align the above along with your fusion transcript sequence and examine the alignment results in the context of the fusion transcript alignments results.
How do the genomic breakpoints compare to the fusion-transcript breakpoints? Why do they differ?
Detection of foreign transcripts (eg. oncoviruses)
Search the de novo reconstructed transcripts for evidence of viruses using the Centrifuge software along with the database of viral sequences provided.
% centrifuge -x centrifuge_VirusDB/abv -f \
-U discasm/trinity_out_dir/Trinity.fasta \
> centrifuge.matches.txt
The above will identify transcripts with sequence matches to known viruses, with the matches provided in the ‘centrifuge.matches.txt’ file, and summarizes the findings in a file ‘centrifuge_report.tsv’.
Examine the ‘centrifuge_report.tsv’ file. Were there any matches?
% cat centrifuge_report.tsv
name taxID taxRank genomeSize numReads numUniqueReads abundance
Bos taurus polyomavirus 1 1891754 species 4697 11 11 1
Remember, this was a cancer cell line RNA-Seq data set. Why might we find the above polyomavirus in such a sample? (Try using Google with terms ‘Bos taurus polyomavirus cell culture’ and see what you find)
Concluding remarks
Congratulations on finishing the DISCASM / GMAP-fusion tutorial! This was, of course, a fairly contrived example of how one might use these tools to explore the somewhat ‘dark matter of the cancer transcriptome’. The data sets used here were small enough to be run through on a basic laptop computer in a short amount of time. Running the above system on larger cancer data sets is far more time consuming and computationally challenging, but highly rewarding with respect to biological discoveries.
Be sure to visit our Trinity CTAT site and subscribe to our Trinity CTAT Google forum to stay apprised of our latest developments and announcements.