High-throughput Biology - From Sequence to Networks 2019 - Lab 5
This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License. This means that you are able to copy, share and modify the work, as long as the result is distributed under the same license.
CSHL-CBW HT-Bio Module 5 - Structural Variant Calling
by Mathieu Bourgey, Ph.D
Table of contents
- Introduction
- Original Setup
- Align DNA with BWA-MEM
- Characterize the fragment size distribution
- Run DELLY to detect SVs
- Setting up IGV for SV visualization
- Explore the SVs
- Acknowledgements
Introduction
The goal of this practical session is to identify structural variants (SVs) in a human genome by identifying both discordant paired-end alignments and split-read alignments that.
Discordant paired-end alignments conflict with the alignment patterns that we expect (i.e., concordant alignments) for the DNA library and sequencing technology we have used.
For example, given a ~500bp paired-end Illumina library, we expect pairs to align in F/R orientation and we expect the ends of the pair to align roughly 500bp apart. Pairs that align too far apart suggest a potential deletion in the DNA sample’s genome. As you may have guessed, the trick is how we define what “too far” is — this depends on the fragment size distribution of the data.
Split-read alignments contain SV breakpoints and consequently, then DNA sequences up- and down-stream of the breakpoint align to disjoint locations in the reference genome.
We will be working on a 1000 genome sample, NA12878. You can find the whole raw data on the 1000 genome website: http://www.1000genomes.org/data
The dataset comes from the Illumina Platinum Genomes Project, which is a 50X-coverage dataset of the NA12891/NA12892/NA12878 trio. The raw data can be downloaded from the following URL.
NA12878 is the child of the trio while NA12891 and NA12892 are her parents.
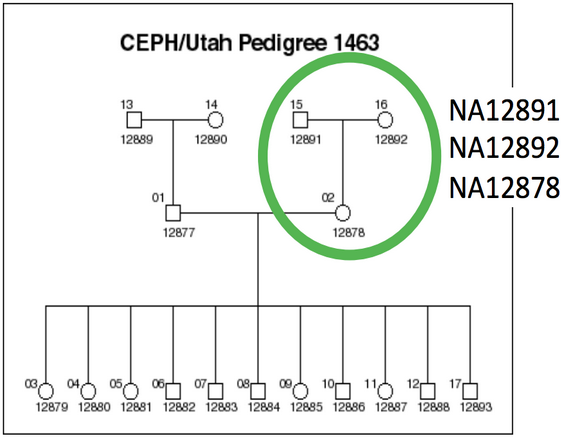
For practical reasons we subsampled the reads from the sample because running the whole dataset would take way too much time and resources. We’re going to focus on the reads extracted from the chromosome 20.
Original Setup
Amazon node
Read these directions for information on how to log in to your assigned Amazon node.
Software requirements
These are all already installed, but here are the original links.
In this session, we will particularly focus on DELLY, a SV detection tool. DELLY is an integrated structural variant prediction method that can discover, genotype and visualize deletions, tandem duplications, inversions and translocations at single-nucleotide resolution in short-read massively parallel sequencing data. It uses paired-ends and split-reads to sensitively and accurately delineate genomic rearrangements throughout the genome.
If you are interested in DELLY, you can read the full manuscript here.
Environment setup
#set up
export WORK_DIR=~/workspace/HTseq/Module5/
export REF=$WORK_DIR/reference
rm -rf $WORK_DIR
mkdir -p $WORK_DIR/SVvariants
cd $WORK_DIR
ln -s ~/CourseData/HT_data/Module5/* .
Note:
The ln -s command adds symbolic links of all of the files contained in the (read-only) ~/CourseData/HT_data/Module5 directory.
Data files
The initial structure of your folders should look like this:
ROOT
|-- bam/ # bam files (down sampled)
`-- NA12878/ # Child sample directory
`-- NA12891/ # Father sample directory
`-- NA12892/ # Mother sample directory
`-- reference/ # hg19 reference and indexes
`-- scripts/ # command lines scripts
`-- saved_results/ # precomputed final files
Cheat sheets
Align DNA with BWA-MEM
This step has been done for you in the interest of time, but the commands are shown so that you can reproduce the results later. The advantage of using BWA-MEM in the context of SV discovery is that it produces both paired-end and split-read alignments in a single BAM output file. In contrast, prior to BWA-MEM, one typically had to use two different aligners in order to produce both high quality paired-end and split-read alignments.
In the alignment commands, note the use of the -M parameter to mark shorter split hits as secondary.
#################
#Align NA12878 #
#################
#bwa mem $REF/hg19.fa \
#fastq/NA12878_S1.chr20.20X.1.fq \
#fastq/NA12878_S1.chr20.20X.2.fq \
#-M -t 2| samtools view -S -b - \
#> bam/NA12878/NA12878_S1.chr20.20X.pairs.readSorted.bam
#################
#Align NA12891 #
#################
#bwa mem hg19.fa \
#fastq/NA12891_S1.chr20.20X.1.fq \
#fastq/NA12891_S1.chr20.20X.2.fq \
#-M -t 2 | samtools view -S -b - \
#> bam/NA12891/NA12891_S1.chr20.20X.pairs.readSorted.bam
#################
#Align NA12892 #
#################
#bwa mem hg19.fa \
#fastq/NA12892_S1.chr20.20X.1.fq \
#fastq/NA12892_S1.chr20.20X.2.fq \
#-M -t 2 | samtools view -S -b - \
#> bam/NA1289/NA12892_S1.chr20.20X.pairs.readSorted.bam
Why should we mark shorter split hits as secondary ? solution
Characterize the fragment size distribution
Before we can attempt to identify structural variants via discordant alignments, we must first characterize the insert size distribution
What means the insert size distribution ? solution
How can we use the fragment size distribution in SV detection ? solution
The following script, taken from LUMPY extracts F/R pairs from a BAM file and computes the mean and stdev of the F/R alignments. It also generates a density plot of the fragment size distribution.
Let’s calculate the fragment distribution for the three dataset:
#NA12878
samtools view bam/NA12878/NA12878_S1.chr20.20X.pairs.readSorted.bam \
| python scripts/pairend_distro.py \
-r 101 \
-X 4 \
-N 10000 \
-o SVvariants/NA12878_S1.chr20.20X.pairs.histo \
> SVvariants/NA12878_S1.chr20.20X.pairs.params
#NA12891
samtools view bam/NA12891/NA12891_S1.chr20.20X.pairs.readSorted.bam \
| python scripts/pairend_distro.py \
-r 101 \
-X 4 \
-N 10000 \
-o SVvariants/NA12891_S1.chr20.20X.pairs.histo \
> SVvariants/NA12891_S1.chr20.20X.pairs.params
#NA12892.
samtools view bam/NA12892/NA12892_S1.chr20.20X.pairs.readSorted.bam \
| python scripts/pairend_distro.py \
-r 101 \
-X 4 \
-N 10000 \
-o SVvariants/NA12892_S1.chr20.20X.pairs.histo \
> SVvariants/NA12892_S1.chr20.20X.pairs.params
-r specifies the read length
-X specifies the number of stdevs from mean to extend
-N specfies the number of read to sample”
-o specifies the output file
Let’s take a peak at the first few lines of the histogram file that was produced:
head -n 10 SVvariants/NA12878_S1.chr20.20X.pairs.histo
Expected results:
0 0.0
1 0.000200461060439
2 0.000300691590659
3 0.000300691590659
4 0.000200461060439
5 0.000200461060439
6 0.000300691590659
7 0.00010023053022
8 0.00010023053022
9 0.00010023053022
Let’s use R to plot the fragment size distribution.
First, launch R from the command line.
R
Now, within R, execute the following commands:
size_dist <- read.table('SVvariants/NA12878_S1.chr20.20X.pairs.histo')
pdf(file = "SVvariants/fragment.hist.pdf")
layout(matrix(1:3))
plot(size_dist[,1], size_dist[,2], type='h', main="NA12878 insert size")
size_dist <- read.table('SVvariants/NA12891_S1.chr20.20X.pairs.histo')
plot(size_dist[,1], size_dist[,2], type='h', main="NA12891 insert size")
size_dist <- read.table('SVvariants/NA12892_S1.chr20.20X.pairs.histo')
plot(size_dist[,1], size_dist[,2], type='h', main="NA12892 insert size")
dev.off()
quit("no")
At this point, you should have the following files:
Let’s look at the 3 PDF files :
open a web browser on your laptop, and navigate to http://XX.oicrcbw.ca, where XX is the id of your node. You should be able to find there the directory hierarchy under ~/workspace/ on your node. open HTseq/Module5/SVvariants folder and open the pdf fragment.hist.pdf
Spend some time thinking about what this plot means for identifying discordant alignments.
What does the mean fragment size appear to be? Are all 3 graphs the same? solution
SV detection
For germline SVs, calling is done by sample or in small batches to increase SV sensitivity & breakpoint precision.
Here are the steps adapted from the DELLY readme.
First call
Let’s call SVs:
#NA12878
delly call -g $REF/hg19.fa -o SVvariants/NA12878.bcf -x $REF/hg19.excl bam/NA12878/NA12878_S1.chr20.20X.pairs.posSorted.bam
#NA12891
delly call -g $REF/hg19.fa -o SVvariants/NA12891.bcf -x $REF/hg19.excl bam/NA12891/NA12891_S1.chr20.20X.pairs.posSorted.bam
#NA12892
delly call -g $REF/hg19.fa -o SVvariants/NA12892.bcf -x $REF/hg19.excl bam/NA12892/NA12892_S1.chr20.20X.pairs.posSorted.bam
.bcf files are compressed vcf files. To look at the output you can use bcftools
bcftools view SVvariants/NA12878.bcf | less -S
Cheat: If these commands are taking too long, simply run the command cp saved_results/SVvariants/NA128*[128].bc* SVvariants/
How many variants delly found in each sample ? solution
How many variants by SV type are found in each sample ? solution
Merge calls
We need to merge the SV sites into a unified site list:
delly merge -m 500 -n 1000000 -o SVvariants/sv.bcf -b 500 -r 0.5 SVvariants/NA12878.bcf SVvariants/NA12891.bcf SVvariants/NA12892.bcf
Look at the output:
bcftools view SVvariants/sv.bcf | less -S
How many variants by SV type are found ? solution
What can you notice something different from the individual bcf ? solution
Re-genotype in all samples
We need to re-genotype the merged SV site list across all samples. This can be run in parallel for each sample.
#NA12878
delly call -g $REF/hg19.fa -v SVvariants/sv.bcf -o SVvariants/NA12878.geno.bcf -x $REF/hg19.excl bam/NA12878/NA12878_S1.chr20.20X.pairs.posSorted.bam
#NA12891
delly call -g $REF/hg19.fa -v SVvariants/sv.bcf -o SVvariants/NA12891.geno.bcf -x $REF/hg19.excl bam/NA12891/NA12891_S1.chr20.20X.pairs.posSorted.bam
#NA12892
delly call -g $REF/hg19.fa -v SVvariants/sv.bcf -o SVvariants/NA12892.geno.bcf -x $REF/hg19.excl bam/NA12892/NA12892_S1.chr20.20X.pairs.posSorted.bam
Look at the output:
bcftools view SVvariants/NA12878.geno.bcf | less -S
We now have our good candidate list genotype for each individual.
Merge the new calls
Merge all re-genotyped samples to get a single VCF/BCF using bcftools merge. Also index the resulting file and create vcf file for visualization.
bcftools merge -O b -o SVvariants/merged.bcf SVvariants/NA12878.geno.bcf SVvariants/NA12891.geno.bcf SVvariants/NA12892.geno.bcf
bcftools index SVvariants/merged.bcf
bcftools view SVvariants/merged.bcf > SVvariants/merged.vcf
Do you know how to look at the resulting file? solution
Setting up IGV for SV visualization
Launch IGV and load the merged calls and the germline calls using File -> Load from URL using:
- http://XX.oicrcbw.ca/HTseq/Module5/SVvariants/merged.vcf
Note: Once again you will need to replace XX by your student number.
Navigate to the following location to see a deletion: chr20:31,308,410-31,315,294
Now load the bam files in the same way using:
- http://XX.oicrcbw.ca/HTseq/Module5/bam/NA12878/NA12878_S1.chr20.20X.pairs.posSorted.bam
- http://XX.oicrcbw.ca/HTseq/Module5/bam/NA12891/NA12891_S1.chr20.20X.pairs.posSorted.bam
- http://XX.oicrcbw.ca/HTseq/Module5/bam/NA12892/NA12892_S1.chr20.20X.pairs.posSorted.bam
You should see something like this:
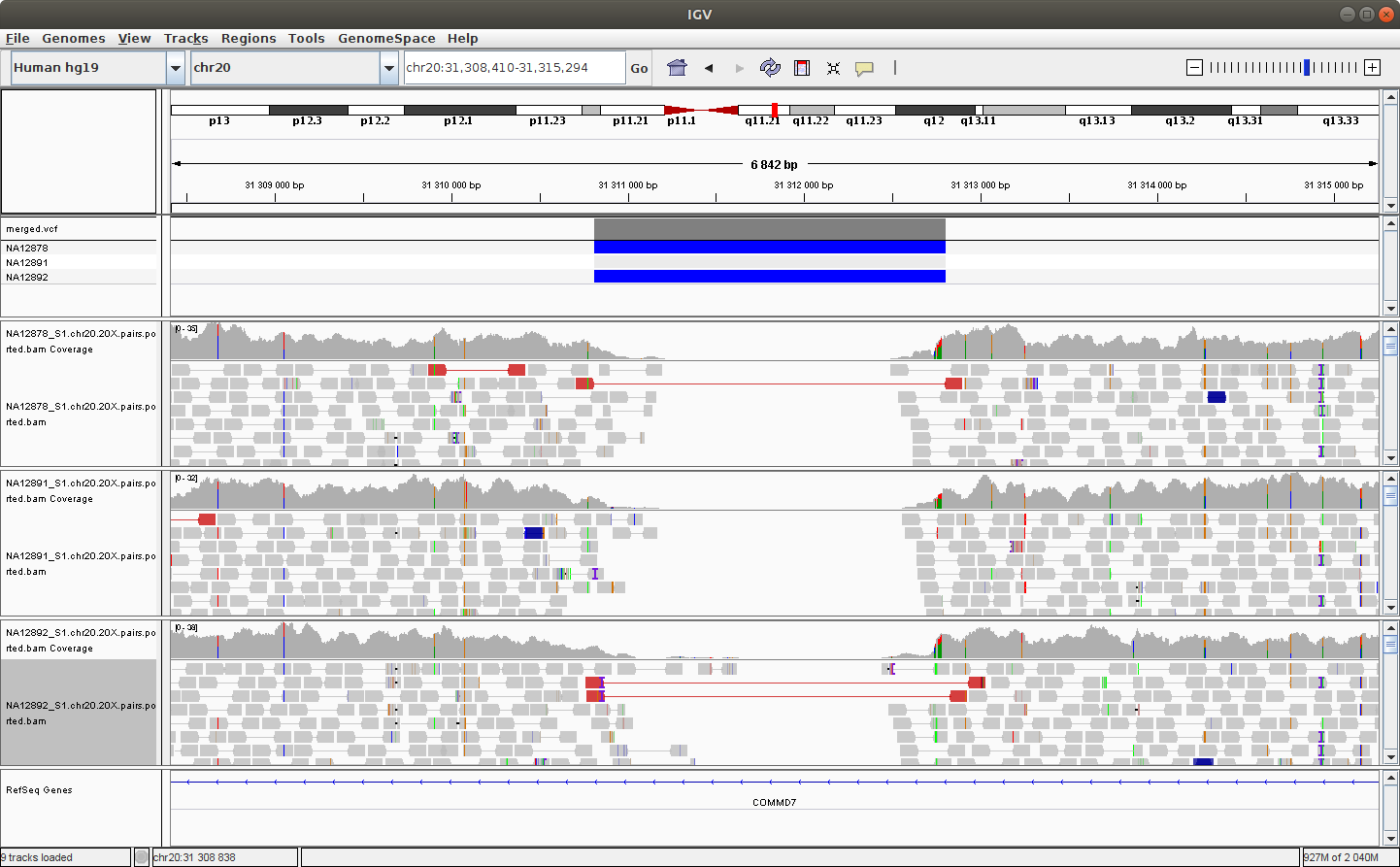
You can try to configure IGV such that we can more clearly see the alignments that support the SV prediction.
Note - Do you remember how to make sure that the alignments are colored by insert size and orientation?*
Explore the SVs
Is the variant at chr20:31,310,769-31,312,959 found in each member of the trio? solution
What are the genotypes for each member of the trio at the locus chr20:61,721,523-61,728,495 (e.g., hemizygous, homozygous)? solution
What about the variant at chr20:52,632,182-52,664,108? solution
Now load the bam files in
- http://XX.oicrcbw.ca/HTseq/Module5/saved_results/Moleculo_bam/NA12878.molelculo.chr20.bam
Does the evidence in the Moleculo track mimic the evidence in the Illumina track for NA12878? solution
What about chr20:18,953,476-18,957,998? solution
Continue exploring the data!
Acknowledgements
This module is heavily based on a previous module prepared by Aaron Quinlan and Guillaume Bourque.