Downstream Analysis and Online Tools
Module 5: Downstream analyses & integrative tools
by David Bujold, M.Sc.
Extra Lab: Galaxy
We will now explore and learn how to use the Galaxy interface. In this short exercise, we will load a FASTQ dataset, run FastQC on it, and trim it to improve overall quality of reads.
- For this exercise, we will use the main Galaxy server. Using a web browser, open the following URL: https://usegalaxy.org/
The types of jobs you can run without registering on the main Galaxy instance are limited. We should therefore create an account.
-
On the top menu, click on “Login or Register”.
-
Click on “Don’t have an account? Register here.”, fill your account information, and validate your email address.
-
Once you are logged in as a Galaxy user, you’re ready to go. For this exercise, we’ll use subsets of data from the Illumina BodyMap 2.0 project, from human adrenal gland tissues. The sampled reads are paired-end 50bp that map mostly to a 500Kb region of chromosome 19, positions 3-3.5 million (chr19:3000000:3500000). (source: https://usegalaxy.org/u/jeremy/p/galaxy-rna-seq-analysis-exercise)
-
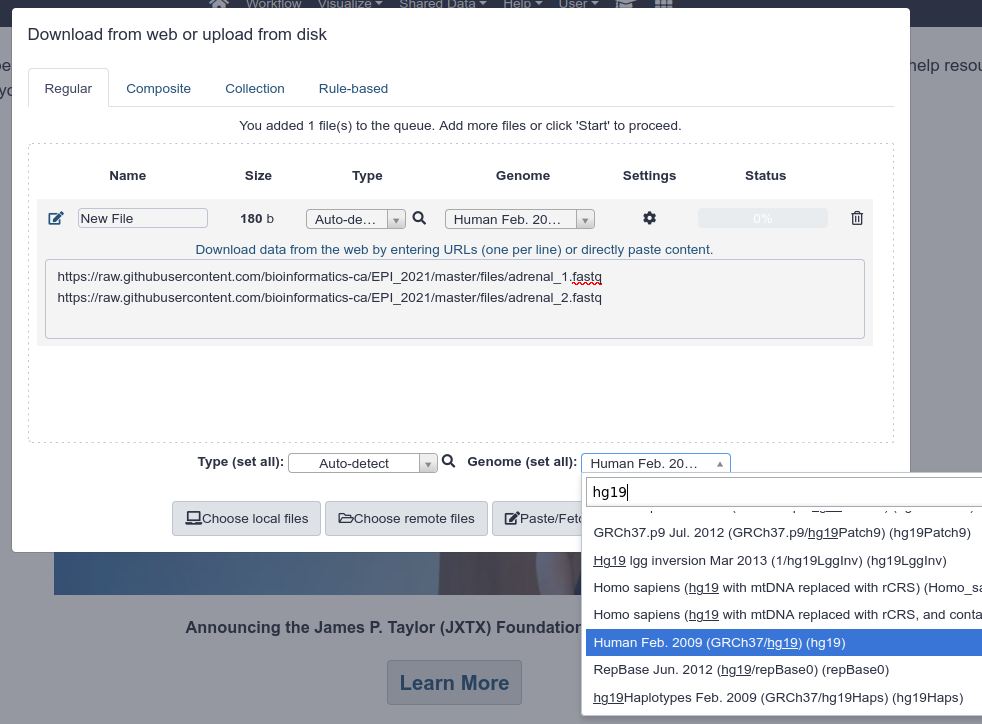
Import the following two FASTQ files in your user space. To do so:
- Click the
Upload Databutton on the upper left of the screen - In the popup, click on the
Paste/Fetch databutton - You can provide both URLs in the same text box
- Under
Genome (set all), specifyHuman Feb. 2009 (GRCh37/hg19) (hg19) - Click on
Start.
- Click the
- After it finished uploading (green state), you can rename the two imported files, for better organization.
- From the history column, click on the
Penicon for the first imported item. Note that you can enter a new name to replace “adrenal_1.fastq” in the dialog if desired. - Examine the adrenal_1.fastq file content using the
Eyeicon.
- From the history column, click on the
- Run the tool FastQC: Comprehensive QC for adrenal_1.
- To find it, use the search window at the top of the Tools column (left panel).
- From the FastQC tool interface, for the field
Raw read data from your current history, choose adrenal_1. - Click on
Run Tool. - Pay attention to the green notice, which provides details about the input and output of the job you just launched.
- Once the job is completed, examine the Webpage results from the history bar using the
Eyeicon. - Raw output statistics are also available, and can also be seen with the
Eyeicon. - Repeat the same operations for adrenal_2. As a shortcut, you can click on the FastQC history item, then click on the
Run this job againicon and simply change the input file to automatically reuse the same parameters.
- If desired, you can run a sanity check on your FASTQ files in Galaxy, to ensure they meet the expected standards. To groom our FASTQ files, we will use the tool
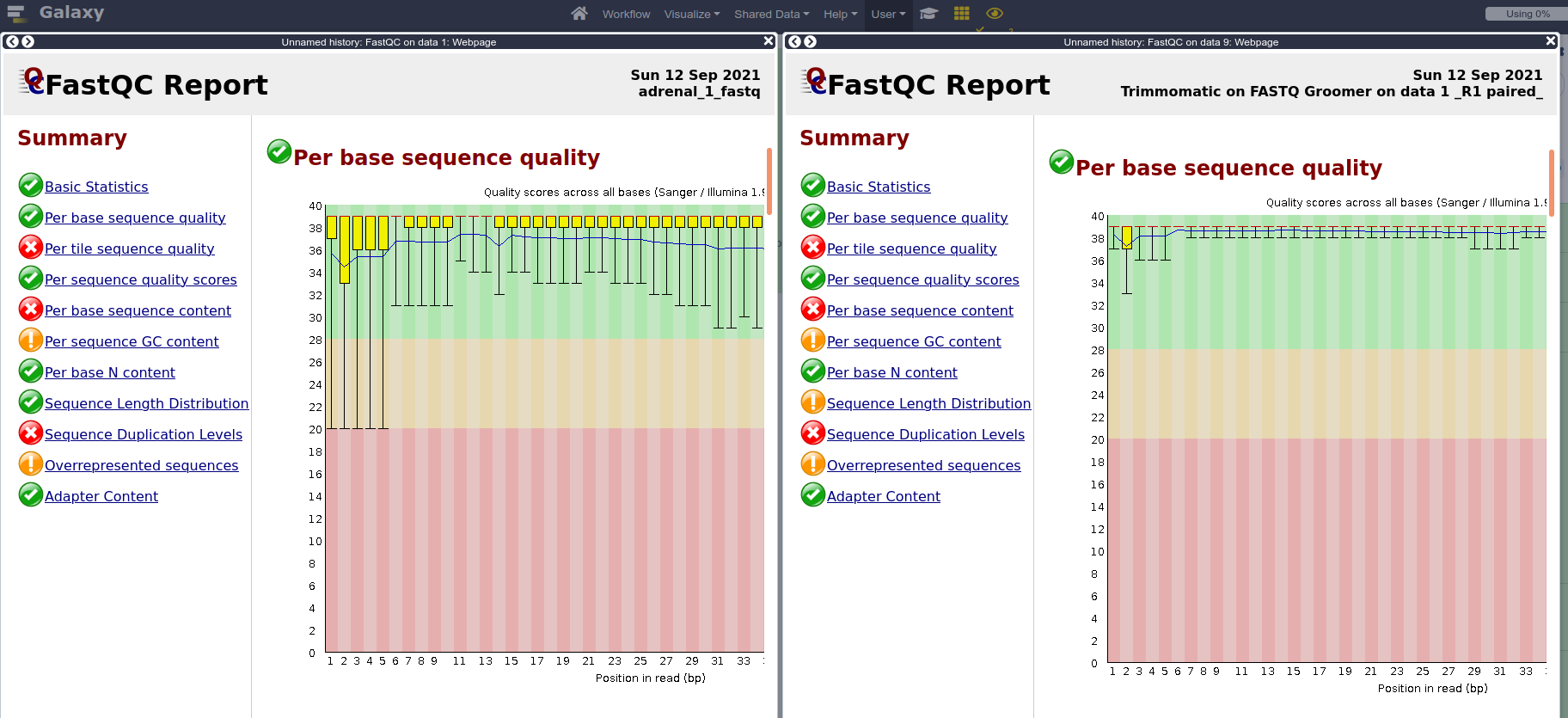
FASTQ Groomerwith default parameters.- When we don’t know which quality score type to provide, we can extract that information from the FastQC report that we already generated. Can you find the information in the FastQC report? (Answer: It’s in Sanger format)
- Leave the other options as-is.
- Run this for both of our paired-end files, adrenal_1 and adrenal_2.
- You will now trim the reads, to improve the quality of the dataset by removing bad quality bases, clipping adapters and so on. Launch the Trimmomatic tool with default parameters, except:
- Set the input as
Paired-end (two separate input files) - Give the groomed adrenal_1 file for direction 1, and groomed adrenal_2 for direction 2.
- Sliding window size: 4
- Average quality required: 30
- Set the input as
- Run FastQC again on both paired files, and compare results with pre-trimming FastQC output.
- As an exercise after the workshop, or if you have time remaining, you can now try to use this dataset in one of the aligners provided by Galaxy, such as HISAT2 or RNA STAR. Once the alignment is completed, try
- downloading the BAM using the
floppy diskicon and visualizing it in IGV - visualizing it in the UCSC Genome Browser, using the
display at UCSC mainoption - creating a bigWig coverage track using the bamCoverage tool
- downloading the BAM using the
