IDE Module 5 Lab
Table of contents
- Learning Objectives
- Introduction
- Using CARD
- AMR Detection from Unassembled Reads
- AMR Detection from Assembled Contigs
- hAMRonizing AMR Detection Results
- Predicting Resistance Phenotype
Learning Objectives
By the end of this practical you will be able to:
-
Use the Comprehensive Antibiotic Resistance Database (CARD) to find information about AMR genes using the Antibiotic Resistance Ontology (ARO).
-
Identify AMR genes in unassembled bacterial reads using the Resistance Gene Identifier’s (RGI) read-mapping mode
-
Assemble bacterial genome reads into contigs and use RGI to identify AMR genes
-
Use hAMRonization to compare and report the results of AMR gene detection in a standardised format
-
Understand the difficulties of predicting phenotype from AMR genomics.
Introduction
The relationship between AMR genotype and AMR phenotype is complicated in-vivo and highly dependent on many factors such as the the composition and spatial organisation of the microbial community and nutrient availability. This means predicting phenotype from genome data alone is very difficult (although probabilistic models can be very helpful and effective especially incorporating additional data). However, genomic (or metagenomic) data is still very useful as it can be used to catalogue the resistome (set of genes and mutations linked to AMR) of an organism or microbial community. This data can then be used to investigate how AMR genes are evolving and spreading in a given outbreak (or more widely), which genes are most likely responsible for observed resistance, and which antimicrobials are more likely to be effective. To ideally profile the resistome we have to compare our genomic or metagenomic data against a specialised database using specialised tools that are designed to detect a range of AMR mechanisms e.g., presence of a homologue, presence of protein or rRNA variants, presence of multi-component systems etc.
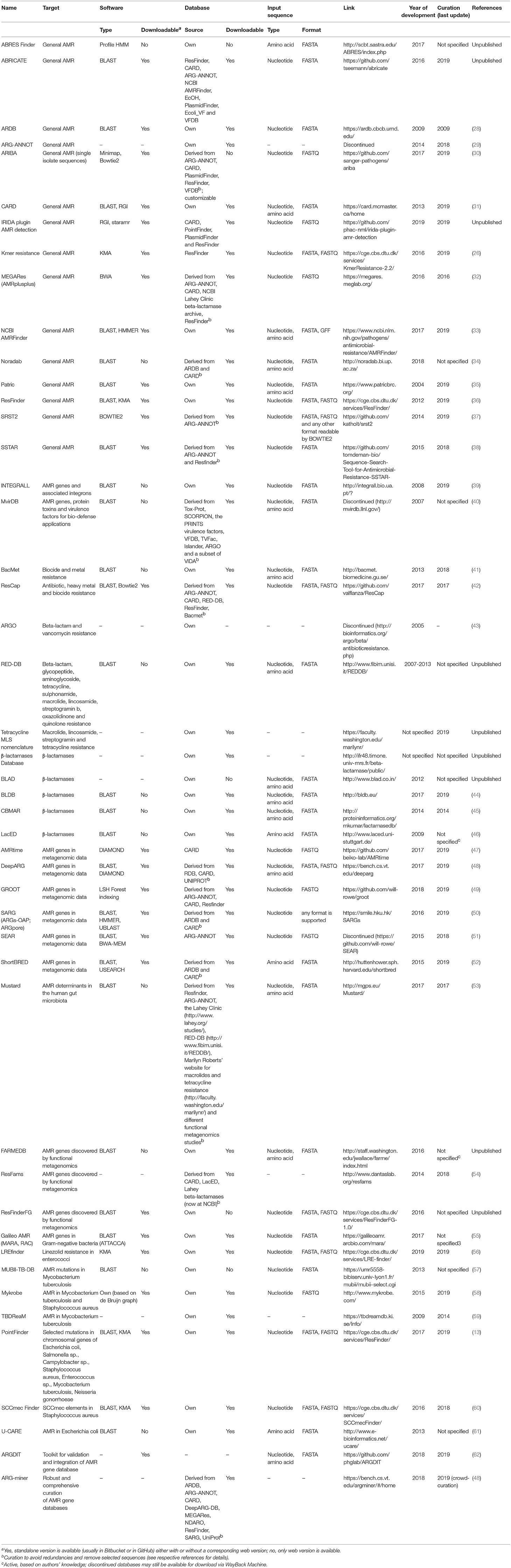
There are several databases (see here for a list) which try and organise information about AMR as well as helping with interpretation of resistome results. Many of these are either specialised on a specific type of resistance gene (e.g., beta-lactamases), organism (e.g., Mycobacterium tuberculosis), or are an automated amalgamation of other databases (e.g., MegaRes). There are also many tools for detecting AMR genes each with their own strengths and weaknesses (see this paper for a non-comprehensive list of tools!).
{kind=link}
The “big 3” databases that are comprehensive (involving many organisms, genes, and types of resistance), regularly updated, have their own gene identification tool(s), and are carefully maintained and curated are:
- Comprehensive Antibiotic Resistance Database (CARD) with the Resistance Gene Identifier (RGI)
- National Center for Biotechnology Information’s National Database of Antibiotic Resistant Organisms (NDARO) with AMRFinderPlus
- ResFinder database with its associated ResFinder tool
There are also both manual and automated efforts to try to ensure co-ordination of metadata and curation between these databases.
In this practical we are going to focus on CARD and the associated RGI tool because A) The Antibiotic Resistance Ontology it is built upon is a great way to organise information about AMR and B) We are biased (CARD is Canadian and pretty much all the CBW faculty collaborate (or are part of) the group that develops CARD)!
Using CARD
To start this tutorial, we will use CARD to examine the diversity of resistance mechanisms, how they influence bioinformatics analysis approaches, and how CARD’s Antibiotic Resistance Ontology (ARO) can provide an organizing principle for interpretation of bioinformatics results.
CARD’s website provides the ability to:
- Browse the Antibiotic Resistance Ontology (ARO) and associated knowledge base.
- Browse the underlying AMR gene detection models, reference sequences, and SNP matrices.
- Download the ARO, reference sequence data, and indices in a number of formats for custom analyses.
- Detect AMR genes in a genome using a web-based RGI.
In this part of the tutorial, your instructor will walk you through the following use of the CARD website to familiarize yourself with its resources.
- What are the mechanism of resistance described in the Antibiotic Resistance Ontology?
- Examine the NDM-1 beta-lactamase protein, it’s mechanism of action, conferred antibiotic resistance, it’s prevalence, and it’s detection model.
- Examine the AAC(6’)-Iaa aminoglycoside acetyltransferase, it’s mechanism of action, conferred antibiotic resistance, it’s prevalence, and it’s detection model.
- Examine the fluoroquinolone resistant gyrB for M. tuberculosis, it’s mechanism of action, conferred antibiotic resistance, and it’s detection model.
- Examine the MexAB-OprM efflux complex, it’s mechanism of action, conferred antibiotic resistance, it’s prevalence, and it’s detection model(s).
Answer
1. + antibiotic target alteration [Resistance Mechanism] + antibiotic target replacement [Resistance Mechanism] + antibiotic target protection [Resistance Mechanism] + antibiotic inactivation [Resistance Mechanism] + antibiotic efflux [Resistance Mechanism] + reduced permeability to antibiotic [Resistance Mechanism] + resistance by absence [Resistance Mechanism] 2. NDM-1: Antibiotic Inactivation, beta-lactams carbapenems etc, lots of ESKAPE pathogens, protein homologue model 3. AAC(6')-Iaa: Antibiotic Inactivation, aminogylcosides, _Salmonella enterica_, protein homologue model 4. gyrB: antibiotic target alteration, fluoroquinolones, _Mycobacterium_, protein variant model 5. MexAB-OprM: antibiotic efflux, everything, have to check components but _Pseudomonas_, efflux meta-modelAMR Detection from Unassembled Reads
The diversity of antimicrobial resistance mechanisms requires a diversity of detection algorithms and a diversity of detection limits. CARD’s Resistance Gene Identifier (RGI) currently integrates four CARD detection models: Protein Homolog Model, Protein Variant Model, rRNA Variant Model, and Protein Overexpression Model. Unlike naïve analyses, CARD detection models use hand-curated cut-offs, currently based on BLAST/DIAMOND bitscore cut-offs. Note: many tools won’t detect mutation related AMR at all so be careful when picking tool if this is important to you.
Using these cut-offs RGI assigns a criteria to the AMR genes its detects:
- Perfect: detected gene identically matches the reference gene in CARD
- Strict: detected gene BLAST/DIAMOND bitscore is more than or equal to the curated cut-off for that detection model
- Loose (if run with
--include_loose): potentially questionable hits below curated bitscore cut-offs
RGI was originally designed to only work on assembled genomic data (using BLAST or DIAMOND) but now incudes a feature to run on unassembled reads (using read-mapping approaches).
There are many other features and options in the RGI tool including ways of summarising and comparing results (type rgi --help in the terminal or reading the documentation for all the details).
We are going to use a lot of these features today!
But first, let us set up RGI and then use it to identify AMR genes in unassembled bacterial reads.
Setting-up
Most of the tool’s we will use in this session should have already been installed in a conda environment in your AWS machine image.
Let’s create a folder to contain our results from this practical and activate that conda environment:
mkdir module5
cd module5
conda activate amr
AMR is a fast moving field, to keep up CARD has monthly updates using a combination of auomated literature mining and a team of manual expert curators. Unless you are trying to reproduce an earlier analysis or maintain consistency within a manuscript, you always want to be using the most recent database version available. Therefore, it is always a good idea to check what database has been installed with RGI (or any other tool) before you run it.
Running this command will tell you the database versions currently being used by RGI:
rgi database --all --version
You can then go to the CARD download page to double-check the latest card_canonical database (called “CARD Data” on the download page) and card_variants (called “CARD Prevalence, Resistomes, & Variants data” on the download page).
You shouldn’t need to do this here but if you ever want to load a new database you can either do it manually with rgi load (instructions in the RGI documentation) or via the rgi autoload feature.
So we’ve got our tool, we’ve got our reference database, now we just need input data!
We’ve pre-loaded your instance with data (and some pre-computed results) from some plasmids and 3 different bacterial species: Neisseria gonorrhoeae, Pseudomonas aeruginosa, and Salmonella enterica (all from analyses in this paper).
You can look at this data by typing:
ls ~/CourseData/IDE_data/module5/reads
ls ~/CourseData/IDE_data/module5/contigs
ls ~/CourseData/IDE_data/module5/contigs/neisseria
ls ~/CourseData/IDE_data/module5/contigs/pseudomonas
ls ~/CourseData/IDE_data/module5/contigs/salmonella
If this data isn’t here and you’ve definitely run ~/init.sh & then you can run the following command to get a backup of the data:
cd ~
scp transfer@deivos.research.cs.dal.ca:/home/transfer/module5_CBW.tar.gz .
tar xvf module5_CBW.tar.gz
Then replace the ~/CourseData/IDE_data/module5 part of the paths below in commands with just ~/module5
Using RGI-BWT
We are going to start by picking just a small set of reads derived from an unknown plasmid. While RGI-BWT is pretty fast it would still take a while to run a full set of genomic (or metagenomic reads)!
mkdir rgi_bwt_results
cd rgi_bwt_results
mkdir unknown_plasmid_1
cd unknown_plasmid_1
Take a look at some of the parameters needed to run rgi bwt:
rgi bwt -h
Then we can run rgi bwt using the bowtie2 short read mapper by providing the individual read files and output
rgi bwt --read_one ~/CourseData/IDE_data/module5/reads/unknown_plasmid_1/unknown_plasmid_1_R1.fq.gz --read_two ~/CourseData/IDE_data/module5/reads/unknown_plasmid_1/unknown_plasmid_1_R2.fq.gz --output_file unknown_plasmid_1_read_rgi --threads 4 --aligner bowtie2 --clean
RGI bwt is still in beta and produces a LOT of output files.
However, the file we are most interested in for now is unknown_plasmid_1_read_rgi.gene_mapping_data.txt
If you open this file (either by downloading it and opening it in a spreadsheet program or by viewing it a text editor such as nano unknown_plasmid_1_read_rgi.gene_mapping_data.txt, you can see AMR genes that have been identified in these reads.
- Which AMR genes were found?
- Which AMR gene had the highest % coverage? Remember this for later
Answer
AMR genes found: * MCR-1 (Avg coverage: 45.2%) * MCR-1.5 (Avg coverage: 54%) * MCR-1.11 (Avg coverage: 57.46%) * MCR-1.7 (Avg coverage: 37.7%) * MCR-1.12 (Avg coverage: 80.57%) * MCR-1.8 (Avg coverage: 36.47%) * MCR-1.13 (Avg coverage: 69.13%) * MCR-1.2 (Avg coverage: 86.35%) (highest coverage) * MCR-1.9 (Avg coverage: 51.29%) * MCR-1.6 (Avg coverage: 42.68%) * MCR-1.3 (Avg coverage: 72.57%) * MCR-1.4 (Avg coverage: 39.42%)Note: if viewing in nano, use Ctrl+X to exit, and pressing the N key if asked to save changes.
Now use cd .., create a new directory, and try and repeat this process using reads from unknown_plasmid_2 (~/CourseData/IDE_data/module5/reads/unknown_plasmid_2)
- Which AMR genes were found in plasmid 2?
- Which AMR gene had the highest % coverage? Remember this for later
-
Do you think these plasmids really have so many closely related alleles on them?
- From these results and what you know about assembly, what do you think are the advantages/disadvantages of read-based methods
Answer
Sample of some of the AMR genes found (coverage %): * TEM-126 (29.62%) * CTX-M-42 (10.16%) * TEM-198 (34.26) * CTX-M-88 (20.55%) * TEM-207 (35.42%) * CTX-M-101 (28.08%) * TEM-122 (32.17%) * TEM-214 (32.06%) * TEM-33 (36.82%) * CTX-M-69 (62,67%) Highest coverage: * sul2 (100%) * APH(6)-Id (100%) * dfrA14 (100%) * APH(3'')-Ib (100%) Total: 70 AMR genes found Read-based analyses advantages and disadvantages: * Higher sensitivity (we find as many AMR genes as possible) * Lower specificity (we are more likely to make mistakes when identifying AMR genes) * Incomplete data (we are likely to find fragments of genes instead of whole genes, this can lead to confusion between similar genes) * No genomic context (we don't know where a gene we detect comes from in the genome, is it associated with a plasmid?)AMR Detection from Assembled Contigs
That’s the theory but how do read-based analyses really compare to assembly based analyses?
First, let’s assemble the same set of reads we just analysed into contigs.
For this we are going to use shovill as it is a convenient way to get quick and reasonable good assemblies in one go.
Again, this is a relatively quick process in the grand scheme for a real set of bacterial genomic reads (5-10 million short-reads typically) but ~10-15 minutes is a lot of time for a tutorial.
Therefore, we are still going to keep using our small plasmid datasets.
Let’s get back to our workfolder and create a result folder for this part of the exercise
cd ../..
mkdir rgi_assembly_results
cd rgi_assembly_results
Then we can run shovill:
shovill --R1 ~/CourseData/IDE_data/module5/reads/unknown_plasmid_1/unknown_plasmid_1_R1.fq.gz --R2 ~/CourseData/IDE_data/module5/reads/unknown_plasmid_1/unknown_plasmid_1_R2.fq.gz --outdir unknown_plasmid_1_assembly --cpus 4
This should result in an assembled contig file to be created in unknown_plasmid_1_assembly/contig.fa.
RGI can then be run using these contigs as input. This will involve RGI automatically detecting genes in the contigs (using prodigal) and comparing those genes against the CARD database using either BLAST or DIAMOND.
We’ll use DIAMOND here as it faster than BLAST (at the cost of a very slightly decreased accuracy).
rgi main -h
rgi main --input_sequence unknown_plasmid_1_assembly/contigs.fa --alignment_tool diamond --num_threads 4 --output_file unknown_plasmid_1_contig_rgi --clean
The output file we are interested in will be called unknown_plasmid_1_contig_rgi.txt
- What AMR genes have been detected?
- How does this compare with the results from RGI-bwt? Is this the same gene that had the most coverage in RGI-bwt?
- Can you use these results with the CARD website to tell me what antibiotics this organism is most likely to be resistant to?
- Using the website can you tell what species this gene is common in proportionally?
Answer
AMR gene found (best hit): MCR-1.2 Significantly smaller list of AMR genes found. Only one gene found, Perfect hit (sequence identity = 100%). Matches the same gene that had the most ccverage in RGI-bwt. From the CARD website, most likely to be resistant tc antibiotics in the peptide antibiotic drug class. Examples include colistin A and colistin B.Repeat this process of assembling and running RGI on the contigs using reads from unknown_plasmid_2 (~/CourseData/IDE_data/module5/reads/unknown_plasmid_2).
- How do these genes compared to the read results from RGI-bwt for plasmid 2?
- Download the
.jsonfile produced by this second run (e.g.,unknown_plasmid_2_contig_rgi.jsondependening on the--output_fileoption you used) and upload it to analyze json option at the bottom of this page (Note: this was broken yesterday so we may have to skip)
Answer
AMR genes found: * MCR-1.1 (Perfect hit) * CTX-M-55 (Perfect) * TEM-1 (Perfect_ * APH(6)-Id (Strict hit) (detected as 100% coverage in RGI=bwt) * APH(3'')-Ib (Strict) (detected as 100% coverage in RGI-bwt) * sul2 (Strict) (detected as 100% coverage in RGI-bwt) * tet(A) (Strict) * dfrA14 (Perfect) (detected as 100% coverage in RGI-bwt)These are only small datasets, we are going to try and run this on a few real genomes later on so remember the commands!
hAMRonizing AMR Detection Results
You’ve done some manual inspect to compare to the two runs but is there a way to make this easier? As mentioned in the lecture we’ve developed a method called “hAMRonization” that allows you to compare results from many different AMR gene detection tools. This requires a few extra pieces of metadata to work that the tool can’t find in the RGI output files:
So let’s try it.
First activate the environment:
cd ..
conda activate hamronization
Then we want to convert the outputs from RGI into this hAMRonized format compatible with every other tool.
# unknown_plasmid_1
hamronize rgi rgi_assembly_results/unknown_plasmid_1_contig_rgi.txt --analysis_software_version 5.2.0 --reference_database_version 3.1.3 --input_file_name unknown_plasmid_1 --output unknown_plasmid_1_contig_hamronize.tsv
hamronize rgi rgi_bwt_results/unknown_plasmid_1/unknown_plasmid_1_read_rgi.gene_mapping_data.txt --analysis_software_version 5.2.0 --reference_database_version 3.1.3 --input_file_name unknown_plasmid_1 --output unknown_plasmid_1_reads_hamronize.tsv
# unknown_plasmid_2
hamronize rgi rgi_assembly_results/unknown_plasmid_2_contig_rgi.txt --analysis_software_version 5.2.0 --reference_database_version 3.1.3 --input_file_name unknown_plasmid_2 --output unknown_plasmid_2_contig_hamronize.tsv
hamronize rgi rgi_bwt_results/unknown_plasmid_2/unknown_plasmid_2_read_rgi.gene_mapping_data.txt --analysis_software_version 5.2.0 --reference_database_version 3.1.3 --input_file_name unknown_plasmid_2 --output unknown_plasmid_2_reads_hamronize.tsv
Finally, we want to create a summary of these hAMRonized results:
# unknown_plasmid_1
hamronize summarize --summary_type interactive --output unknown_plasmid_1_comparison.html unknown_plasmid_1_contig_hamronize.tsv unknown_plasmid_1_reads_hamronize.tsv
# unknown_plasmid_2
hamronize summarize --summary_type interactive --output unknown_plasmid_2_comparison.html unknown_plasmid_2_contig_hamronize.tsv unknown_plasmid_2_reads_hamronize.tsv
You can then download the unknown_plasmid_1_comparison.html and unknown_plasmid_2_comparison.html files and open it in your browser to see an interactive comparison of your results.
Download the new interactive summary and compare the results for the two plasmids in contig and bwt modes:
- Based on this comparison and the advantages/disadvantages of read-based methods, what are the advantages/disadvantages of contig-based analyses?
Answer
Advantages/disadvantages of contig based methods: * Potentially lower sensitivity (we may not find all the AMR genes present due to data loss in assembly) * Higher specificity (we are more likely to identify the correct AMR gene) * More complete data (we are likely to find less fragmentary data with a decent assembly!) * Genomic context (we can find out what genes are next to the AMR gene!)Summarising Resistome Data
RGI has built-in tools to visualize the resistance genes found across many samples including potential phenotypic resistance profiles and AMR genes families in the form of heatmaps.
Earlier in this lab, we mentioned pre-loaded data from 3 different bacterial species: let’s compare the resistance profile between each of them.
For review:
ls ~/CourseData/IDE_data/module5/contigs
ls ~/CourseData/IDE_data/module5/contigs/neisseria
ls ~/CourseData/IDE_data/module5/contigs/pseudomonas
ls ~/CourseData/IDE_data/module5/contigs/salmonella
From each of the species directories, you are free to select one sample of your choice for the following exercise, but for this demo, the samples we will use are the following:
ls ~/CourseData/IDE_data/module5/contigs/neisseria/SAMD00099400.contigs.fa.gz
ls ~/CourseData/IDE_data/module5/contigs/pseudomonas/SAMD00019033.contigs.fa.gz
ls ~/CourseData/IDE_data/module5/contigs/salmonella/SAMN06286049.contigs.fa.gz
Start by creating another directory to store RGI results from contigs within your module5 directory:
mkdir rgi_results
cd rgi_results
conda activate amr
Review the parameters needed to run rgi main:
rgi main -h
Then using the above path to selected samples, we can run rgi main:
# From Neisseria directory:
rgi main --input_sequence ~/CourseData/IDE_data/module5/contigs/neisseria/SAMD00099400.contigs.fa.gz --alignment_tool diamond --num_threads 4 --output_file neisseria_rgi --clean
# From Pseudomonas directory
rgi main --input_sequence ~/CourseData/IDE_data/module5/contigs/pseudomonas/SAMD00019033.contigs.fa.gz --alignment_tool diamond --num_threads 4 --output_file pseudomonas_rgi --clean
# From Salmonella directory
rgi main --input_sequence ~/CourseData/IDE_data/module5/contigs/salmonella/SAMN06286049.contigs.fa.gz --alignment_tool diamond --num_threads 4 --output_file salmonella_rgi --clean
Note the output files for RGI (neisseria_rgi.txt, pseudomonas_rgi.txt, and salmonella_rgi.txt):
- What AMR genes have been detected? How do they compare across species?
- How do the resistance mechanisms compare (
awk -F$'\t' 'NR>1 {print $16}' pseudomonas_rgi.txt | sort | uniq -c | sort -n -rvsawk -F$'\t' 'NR>1 {print $16}' salmonella_rgi.txt | sort | uniq -c | sort -n -r)
Now let us compare the resistance profile of each species using rgi heatmap.
We can visually compare and contrast the resistance genes and phenotypic resistance between numerous RGI results.
First create another new directory within your module5 directory to store the heatmap results.
mkdir heatmaps
cd heatmaps
Now we can refer to our rgi_results directory when running rgi heatmap which will pull all the .json files.
Each column within the heatmap will correspond to the name of the RGI .json file.
We will generate a few heatmaps across the 3 species categorizing AMR gene families and drug class resistance phenotype
rgi heatmap –h
rgi heatmap -i ~/workspace/module5/rgi_results -cat gene_family -o genefamily_samples -clus samples
rgi heatmap -i ~/workspace/module5/rgi_results -cat drug_class -o drugclass_samples -clus samples
rgi heatmap -i ~/workspace/module5/rgi_results -o cluster_both -clus both
rgi heatmap -i ~/workspace/module5/rgi_results -o cluster_both_frequency -f -clus both
Note for each heatmap, the following:
- Yellow represents a perfect hit
- Teal represents a strict hit
- Purple represents no hit
Download the .png files generated and refer to your answers to the previous set of questions:
- At a glance, can you identify AMR gene families that are found in all three species?
Answer
No AMR gene families seem to be shared between all three species; however, there are some shared between two: * adeF and rsmA genes are shared between _Pseudomonas_ and _Salmonella_ which are part of the resistance-nodulation-cell division (RND) antibiotic efflux pump AMR gene familyNow of course, we only looked at a single sample from each species, so now we will analyse Using pre-compiled RGI results, we can create heatmaps to analyse resistance within a single species using Neisseria gonorrhoeae as an example:
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/neisseria -cat gene_family -o genefamily_neisseria -clus samples
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/neisseria -cat drug_class -o drugclass_neisserias -clus samples
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/neisseria -o cluster_both_neisseria -clus both
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/neisseria -o cluster_both_frequency_neisseria -f -clus both
Feel free to re-run the above adjusting for Pseudomonas aeruginosa and/or Salmonella enterica!
Lastly, we can create a heatmap summarizing the resistance profiles for all 33 pre-compiled samples:
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/all -cat gene_family -o genefamily_all_samples -clus samples
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/all -cat drug_class -o drugclass_all_samples -clus samples
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/all -o cluster_both_all_samples -clus both
rgi heatmap -i ~/CourseData/IDE_data/module5/precompiled_rgi/all -o cluster_both_frequency_all_samples -f -clus both
- Which species may be resistant to the most types of antibiotics?
Answer
_Pseudomonas_ - not only with many strict hits but perfect hits to genes conferring resistance to a number of antibiotics relative to _Salmonella_ or _Neisseria_Relax
You can now identify AMR genes from contigs and reads, then use CARD to interpret the results.
This is the main building block of AMR epidemiology when combined with genome assembly, mobile genetic element analysis, and phylogenetics which you’ll learn in other modules!