Informatics for RNA-Seq Analysis 2019
Intro to Genome-guided RNA-Seq Assembly
To make use of a genome sequence as a reference for reconstructing transcripts, we’ll use the Tuxedo2 suite of tools, including Hisat2 for genome-read mappings and StringTie for transcript isoform reconstruction based on the read alignments. We’ll also explore using Trinity in genome-guided mode, performing a de novo assembly for reads aligned and clustered along the reference genome.
The data set that we’ll be leveraging for this application are human RNA-Seq data corresponding to a subset of genes on the human X chromosome, all based on data provided as part of the Tuxedo2 protocol paper.
This excercise provides a quick introduction to the Tuxedo2 toolkit and leverages only a single small pair of fastq files and a ‘miniaturized’ version of the X chromosome containing ~100 genes. For more detailed exploration of genome-guided assembly, you are encouraged to read the Tuxedo2 protocol paper and leverage the complete data sets involving multiple sample replicates.
Data Overview
The data sets we’ll use for genome-guided assembly are located at:
% ls ~/CourseData/RNA_data/trinity_trinotate_tutorial/mini_humanX/
.
minigenome.fa minigenome.gtf reads_1.fq.gz reads_2.fq.gz
where we have a ‘minigenome.fa’ genome fasta file and corresponding ‘minigenome.gtf’ file providing the corresponding gene structure annotations. A single pair of fastq files is provided as ‘reads_1.fq.gz’ and ‘reads_2.fq.gz’.
How many reads are there?
Setting up your environment
For convenience, we’ll be making use of certain environmental variables, such as $TRINITY_HOME to define where the Trinity software is installed.
To configure your environment, simply run the following command:
% source ~/CourseData/RNA_data/trinity_trinotate_tutorial/environment.txt
Now, if you type
% env | grep TRINITY
you should see:
TRINITY_HOME=/usr/local/trinityrnaseq-Trinity-v2.8.4/
Setting up your workspace
In your workspace directory, create a new workspace called ‘workspace_GG’ (with the ‘GG’ short for genome-guided).
% cd ~/workspace # change to home directory
% mkdir workspace_GG
% cd workspace_GG
Create symbolic links (shortcuts) to our reference genome, annotation, and read files:
% ln -s ~/CourseData/RNA_data/trinity_trinotate_tutorial/mini_humanX/* .
At this point, you’re familiar with FASTA (.fa) files and FASTQ (.fq) files. The .gtf file provides the genome annotation and it’s formatted like so:
% head minigenome.gtf
.
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_172246"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform b precursor"; orig_coord_info "chrX,NM_172246,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_172245"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform a precursor"; orig_coord_info "chrX,NM_172245,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_006140"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform a precursor"; orig_coord_info "chrX,NM_006140,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_001161529"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform a precursor"; orig_coord_info "chrX,NM_001161529,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_001161532"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform h"; orig_coord_info "chrX,NM_001161532,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_001161531"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform g precursor"; orig_coord_info "chrX,NM_001161531,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_172249"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform e precursor"; orig_coord_info "chrX,NM_172249,1268800,1268879,+";
minigenome hg38_refGene exon 1001 1080 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NR_027760"; gene_name "CSF2RA"; orig_coord_info "chrX,NR_027760,1268800,1268879,+";
minigenome hg38_refGene exon 3651 3738 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_172246"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform b precursor"; orig_coord_info "chrX,NM_172246,1274755,1274842,+";
minigenome hg38_refGene exon 3651 3714 0.000000 + . gene_id "CSF2RA"; transcript_id "CSF2RA^NM_172245"; gene_name "CSF2RA"; product "granulocyte-macrophage colony-stimulating factor receptor subunit alpha isoform a precursor"; orig_coord_info "chrX,NM_172245,1274755,1274818,+";
GTF format is a popular way to store genome feature annotations.
Tuxedo2
The Tuxedo2 protocol involves first aligning reads to the genome using hisat2, followed by transcript reconstruction using StringTie.
Aligning reads to the genome using Hisat2
Before we can align reads to the genome, we must index for use with hisat2. This involves a few operations:
Extract splice sites from intron-containing transcript structures:
% hisat2_extract_splice_sites.py minigenome.gtf > minigenome.gtf.ss
Extract the exon records:
% hisat2_extract_exons.py minigenome.gtf > minigenome.gtf.exons
Now build the hisat2 index of the genome, leveraging the splice sites and exon data extracted above:
% hisat2-build --exon minigenome.gtf.exons --ss minigenome.gtf.ss minigenome.fa minigenome.fa
After the genome index has been created, we’re ready to align reads to the genome. The native output from hisat2 is SAM and so we pipe this into samtools to convert to BAM and coordinate-sort the output.
% hisat2 --dta -x minigenome.fa --max-intronlen 5000 \
-1 reads_1.fq.gz -2 reads_2.fq.gz \
| samtools view -Sb \
| samtools sort -o alignments.hisat2.bam
The above command limits the intron length to 5k only because our ‘minigenome’ was constructed to limit intron lengths to a max of 5kb. The minigenome is far more dense than the true human X chromosome.
Examine the contents of the bam file using ‘samtools’:
% samtools view alignments.hisat2.bam | head
.
ERR188245.78785_HWI-ST661:130:C037KACXX:6:1101:8916:31520 137 minigenome 2183 60 4S71M = 2183 0 GTACGCCTGTAATCCCAGTGACTTGGGAGGCTGAGGCAGGAGAATCGCTTGAACCCGGGAGGCGGAGGTTGCAGT C@@FFFFFHHHHHJJJJJGHHIJJJJIIJJJJIIJJIIIJEGHJIIIIIJFGGIJJEHFABDBDD;BB2?CBCC@ AS:i:-14 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:14C0T55 YT:Z:UP NH:i:1
ERR188245.78785_HWI-ST661:130:C037KACXX:6:1101:8916:31520 69 minigenome 2183 0 * = 2183 0 GGATAATCATGTTCTTGATACACATTTCCTTTTTTTTTGAGATGGAGTCTCGCTGTTGCCCAGGCTGGAGTACAG CCCFFFFFHHHHHJJJJIJJJJJJJJJJJJJJJJJJJJBC@FCHCCGC==E9@EAEH:@DFFDDDEDDABCDDDE YT:Z:UP
ERR188245.525513_HWI-ST661:130:C037KACXX:6:1101:18753:196335 73 minigenome 2183 60 1S74M = 2183 0 CGCCTGTAATCCCAGTGACTTGGGAGGCTGAGGCAGGAGAATCGCTTGAACCCGGGAGGCGGAGGTTGCAGTGAG CCCFFFFFHHHHHJJHIJJJJJJJIJJIJIIJJIJJJGGIJGGGGGICFHIJJIIHEDDBDDBBD<AC?CD:AAC AS:i:-12 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:14C0T58 YT:Z:UP NH:i:1
ERR188245.525513_HWI-ST661:130:C037KACXX:6:1101:18753:196335 133 minigenome 2183 0 * = 2183 0 CGCCAACTCTTCTATATAATCAGTTTGATGATCTGAATTAGAAAATACCCCTGGATAATCATGTTCTTGATACAC C@CFFFFFHHHHHJJJJJJJJIJHJJJJJJJJJJGIIGIIJJJJJCIJJJHIJJJJIJJJJJJJJJJIJJJJJIJ YT:Z:UP
ERR188401.42420_HWI-ST0814:100:D0BT5ACXX:4:1101:20732:26752 69 minigenome 2183 0 * = 2183 0 ATCAAGCCAGCCTGACCAACATGGTGAAACCCCATCTGTACTAAACATAAAAAAATTAGCCTGGCATGGTGGTGT @CCFFFFFHHHHHJJJIJJIJJJJHGIIJIIJJJIJJJHFHIIJICHIJIJJIIIJGICEHGHFFFFEE;CD@?B YT:Z:UP
ERR188401.66672_HWI-ST0814:100:D0BT5ACXX:4:1101:3195:41421 73 minigenome 2183 60 75M = 2183 0 GCCTGTAATCCCAGTGACTTGGGAGGCTGAGGCAGGAGAATCGCTTGAACCCGGGAGGCGGAGGTTGCAGTGAGC CCCFFFFFHHHHHJFHJJJJJJJIJJJJJIJJJJIJIIIIGAFDHIGIIIIJJJHEBD?BDBBD8?CD@CACACD AS:i:-10 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:14C0T59 YT:Z:UP NH:i:1
ERR188401.66672_HWI-ST0814:100:D0BT5ACXX:4:1101:3195:41421 133 minigenome 2183 0 * = 2183 0 TTTTGAGATGGAGTCTCGCTGTTGCCCAGGCTGGAGTACAGTGGCGCGATCTTAGCTCACTGCAACCTCCGCCTC CCCFFFFFHHHHHJJJIJJIJHJJJJJJJJJIEBHIBFHIIECHIJJGIGHIIJJHHHEHFFFFFFFEEDDDDDD YT:Z:UP
ERR188401.42420_HWI-ST0814:100:D0BT5ACXX:4:1101:20732:26752 153 minigenome 2183 60 1S74M = 2183 0 CGCCTGTAATCCCAGTGACTTGGGAGGCTGAGGCAGGAGAATCGCTTGAACCCGGGAGGCGGAGGTTGCAGTGAG DDEEDCEEFFFFHHGHECFJIGIGFJHIIHGIGJJJIHGJJIJJJJJJHGJIJJJJJJJJJJHGHHHFFFFFCCC AS:i:-11 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:14C0T58 YT:Z:UP NH:i:1
ERR188428.130590_HWI-ST0814:100:D0BT5ACXX:4:1101:14424:73163 69 minigenome 2185 0 * = 2185 0 AAAAAATTAATGTGTATCCAGGACATTTTAAAAACCTGTACACAGTGTTTATTGTGGTTAGGAAGCAATTTCCCA CCCFFFFFHHHHHJIIJJJJJIJJJJJJJJJJJIHIJJJIJJJIJGGGIJJJJJIJJJJJJJJJJJJJJIHGHHG YT:Z:UP
ERR188428.130590_HWI-ST0814:100:D0BT5ACXX:4:1101:14424:73163 153 minigenome 2185 60 73M2S = 2185 0 CTGTAATCCCAGTGACTTGGGAGGCTGAGGCAGGAGAATCGCTTGAACCCGGGAGGCGGAGGTTGCAGTGAGCGG DDDDDEDDEFFFFFFFHHHHJJIJIJIIJJJJJJJJJJJJJJJJHF@IJJJJJJJJJJJJJJHHGHHFFFFFCCC AS:i:-12 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:12C0T59 YT:Z:UP NH:i:1
What do the different fields represent? Which is the sam flag?
Use the https://broadinstitute.github.io/picard/explain-flags.html utility to decipher the different flag values
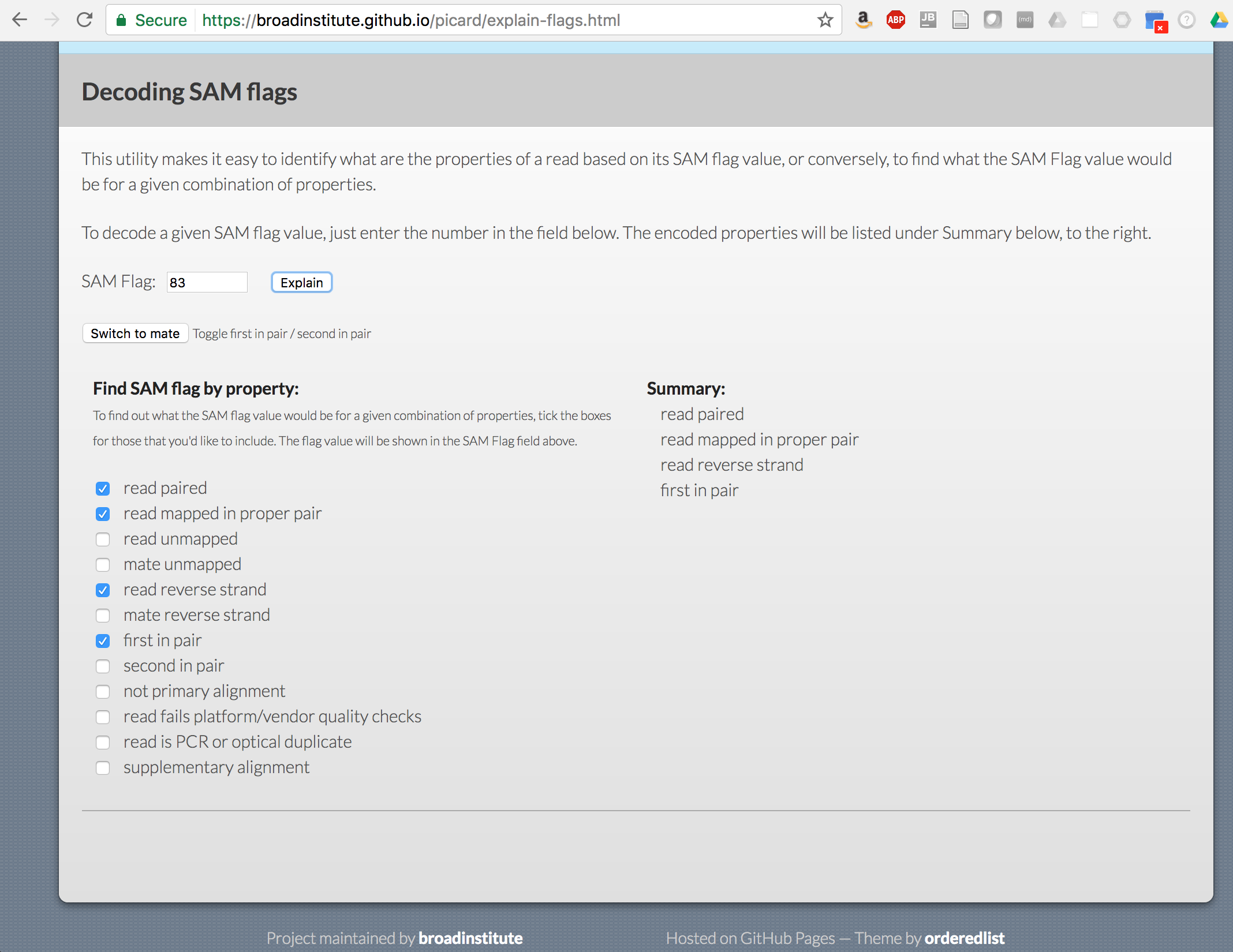
Examine the reads aligned to the genome using IGV. Don’t forget to first index the .bam (to .bai) and .fa (to .fai) files before loading into IGV. (Do you remember the indexing commands for .bam and .fa files? hint: use ‘samtools’). Also load in the reference annotation file ‘minigenome.gtf’ to provide additional perspective.
Transcript reconstruction using StringTie
StringTie reconstructs transcripts from the aligned reads, leveraging the .bam file as input, and generating a GTF file containing transcript structures as output.
% stringtie -o stringtie.gtf alignments.hisat2.bam
Examine the stringtie.gtf output:
% head stringtie.gtf
# stringtie -o stringtie.gtf alignments.hisat2.bam
# StringTie version 1.3.5
minigenome StringTie transcript 36732 41779 1000 - . gene_id "STRG.1"; transcript_id "STRG.1.1"; cov "3.432668"; FPKM "1150.780029"; TPM "2182.717041";
minigenome StringTie exon 36732 37082 1000 - . gene_id "STRG.1"; transcript_id "STRG.1.1"; exon_number "1"; cov "3.341223";
minigenome StringTie exon 41667 41779 1000 - . gene_id "STRG.1"; transcript_id "STRG.1.1"; exon_number "2"; cov "3.716716";
minigenome StringTie transcript 47357 81576 1000 - . gene_id "STRG.2"; transcript_id "STRG.2.1"; cov "9.523120"; FPKM "3192.565186"; TPM "6055.428711";
minigenome StringTie exon 47357 47707 1000 - . gene_id "STRG.2"; transcript_id "STRG.2.1"; exon_number "1"; cov "4.632516";
minigenome StringTie exon 52409 52531 1000 - . gene_id "STRG.2"; transcript_id "STRG.2.1"; exon_number "2"; cov "11.336511";
minigenome StringTie exon 53825 53968 1000 - . gene_id "STRG.2"; transcript_id "STRG.2.1"; exon_number "3"; cov "14.663729";
minigenome StringTie exon 54834 54966 1000 - . gene_id "STRG.2"; transcript_id "STRG.2.1"; exon_number "4"; cov "15.149121";
Notice that in addition to reconstructing transcripts, StringTie also provides expression values in TPM and FPKM values along with read coverage stats for the transcript and individual exons.
Load the stringtie.gtf file into IGV. How do the stringtie transcript structures compare to the reference transcripts?
Genome-guided Trinity de novo assembly
If your RNA-Seq sample differs sufficiently from your reference genome and you’d like to capture variations within your assembled transcripts, you might consider performing a genome-guided de novo assembly. Trinity can accept a bam file containing genome-aligned rna-seq reads as input. Reads are partitioned into coverage groups along the reference genome and each read cluster is assembled using the standard Trinity de novo assembly. Here, de novo assembly is restricted to only those reads that map to the genome. The advantage is that reads that share sequence in common but map to distinct parts of the genome will be targeted separately for assembly. The disadvantage is that reads that do not map to the genome will not be incorporated into the assembly. Unmapped reads can, however, be targeted for a separate genome-free de novo assembly.
Trinity assembly from genome-aligned reads (bam file)
Run genome-guided Trinity leveraging our hisat2-aligned reads like so:
% $TRINITY_HOME/Trinity --genome_guided_bam alignments.hisat2.bam \
--CPU 2 --max_memory 1G --genome_guided_max_intron 5000 \
--output trinity_out_dir_GG
Once Trinity completes, you’ll once again a trinity_out_dir_GG/ in your new workspace, and in this case it’ll contain the resulting assembly as ‘trinity-GG.fasta’.
Examine this ‘trinity-GG.fasta’ file:
% less trinity_out_dir_GG/Trinity-GG.fasta
.
>TRINITY_GG_1_c0_g1_i1 len=495 path=[1:0-494] [-1, 1, -2]
AGTTATTCAAGTTGTAAAAGGTTATACAATAATTTAACAACTACCTTTTTTATTCTGTCG
GGTTACTGACCTCACTTTATGTAAATACTTCGCATGACAAATTCAGTAACTCGTCTATTT
CAGCATGCATAAGACTTTTCACTAGGGAAACTGATAAAGCTTGAGTCAACTAAATCTGCC
TTCATACTTTATCAAGGGGAACCAAGCCTGCTGTGCTTACATCAGCATCTGGAAGACTTT
CCTCTCCTCTAATCTGTGTACACATCTCCAAGCAAGGAAGAAAAAACAAACTCTGCTCAG
ACGCCTATGAAACACCTGAATGAACTTTGATGAAGTACAGTCTGAGTTACCATCATGCAC
AAGTAGAACTGCTCTTGGACTTGTTTTCCTGTTGTTTGTGGAACCTACGCGTTTGAATGG
CTTGAACGTTGCATCTTTTAAAGTTATTTTTTAAGGGTTCTTGGCATTTATCCTAGTTGT
CCGTGTTTGGCAATG
>TRINITY_GG_2_c0_g1_i1 len=227 path=[1:0-226] [-1, 1, -2]
TAGAGGAGAAAATTTCTATGGTCTAGATATTACTTGTAAAGACATGAGAAACCTGAGGTT
CGCTTTGAAACAGGAAGGCCACAGCAGAAGAGATATGTTTGAGATCCTCACGAGATACGC
GTTTCCCCTGGCTCACAGTCTGCCATTATTTGCATTTTTAAATGAAGAAAAGTTTAACGT
GGATGGATGGACAGTTTACAATCCAGTGGAAGAATACAGGATGCCGG
Aligning Trinity-assembled Transcripts to the Genome
We’ll use the GMAP software to align the Trinity transcripts to our reference genome. Trinity contains a utility that facilitates running GMAP, which first builds an index for the target genome followed by running the gmap aligner:
% ${TRINITY_HOME}/util/misc/process_GMAP_alignments_gff3_chimeras_ok.pl \
--genome minigenome.fa \
--transcripts trinity_out_dir_GG/Trinity-GG.fasta \
--SAM | samtools view -Sb | samtools sort -o trinity-GG.gmap.bam
Index the bam file and import it into IGV to view alongside the aligned reads and the stringtie transcripts.
How do the Trinity-reconstructed transcripts compare to StringTie?
Genome-free de novo assembly
Next, try assembling the reads directly, without using the genome sequence:
% $TRINITY_HOME/Trinity --left reads_1.fq.gz --right reads_2.fq.gz \
--seqType fq --CPU 2 --max_memory 1G \
--output trinity_out_dir
You’ll find the Trinity assembly output file as:
trinity_out_dir/Trinity.fasta
How many transcripts did Trinity reconstruct?
Exploring transcript structures can be more challenging when you do not have a genome sequence to serve as a reference for orienting transcripts and defining intron/exon structures or comparing structures of alternatively spliced variants.
There is a tool called Bandage that allows you to explore your transcriptome assembly, and the various structures that may result from alternative splicing.
Try downloading/installing Bandage, importing your Trinity de novo assembly, and exploring the data. Do you find evidence of alternative splicing within your assembly?
Below is a view of a Trinity assembly from within Bandage.
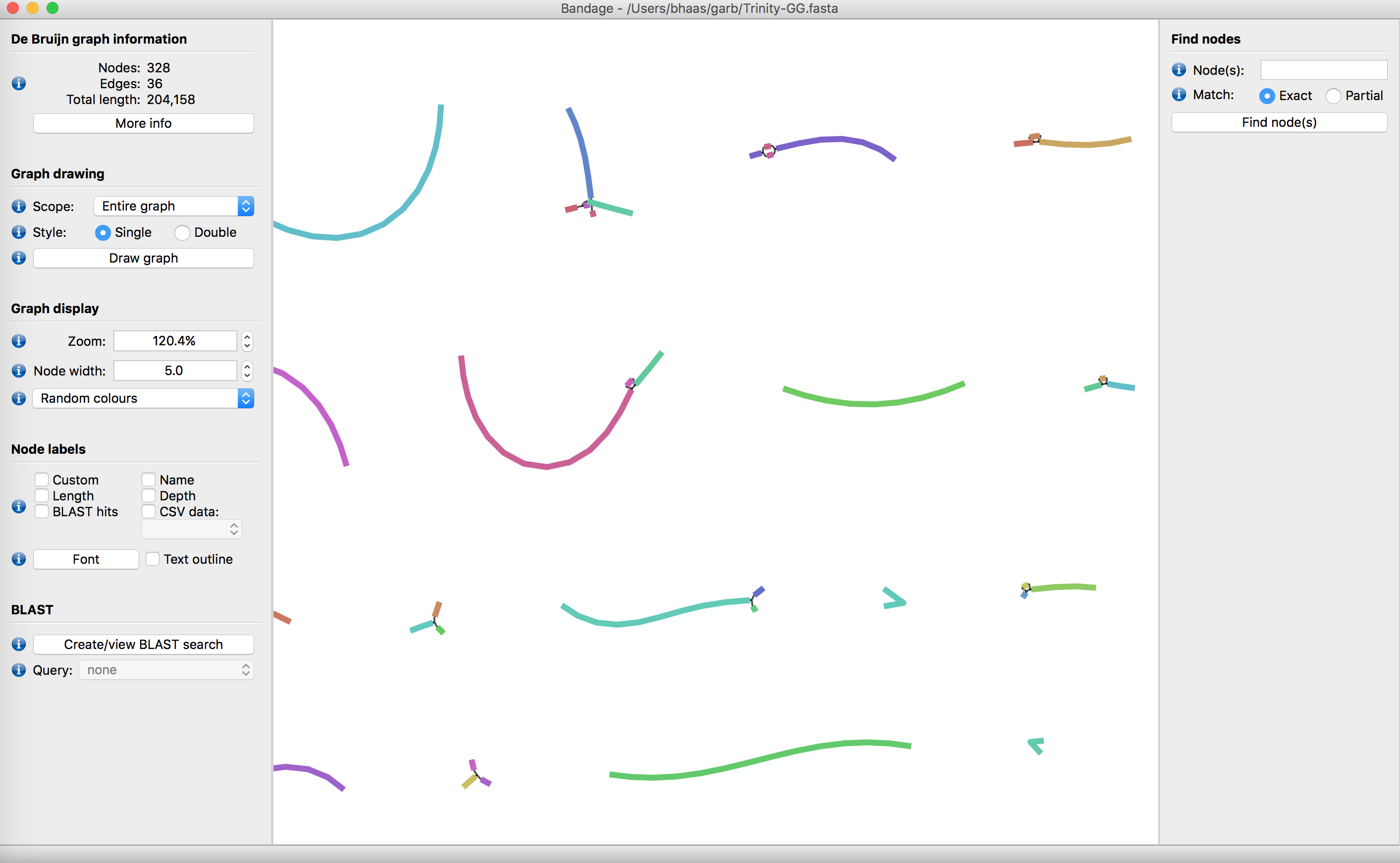
Do you find evidence for alternative splicing?