Lab Module 13 - gProfiler
Lab Module 13 - Pathway Over-representation Analysis
By Jüri Reimand
This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License. This means that you are able to copy, share and modify the work, as long as the result is distributed under the same license.
Let’s get a few gene lists for analysis

Highlights:
- Using the integrated data sets, the authors identified 127 significantly mutated genes as candidate cancer driver genes
- Genes under positive selection, either in individual or multiple tumour types, tend to display higher mutation frequencies above background.
- The statistical analysis identified 127 such genes
- The mutational significance in cancer (MuSiC) package was used to identify significant genes for both individual tumour types and the Pan-Cancer sample cohort. Dees et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012
- These significantly mutated genes are involved in a wide range of cellular processes, including transcription factors/regulators, histone modifiers, genome integrity, receptor tyrosine kinase signalling, cell cycle, mitogen- activated protein kinases (MAPK) signalling, phosphatidylinositol-3-OH kinase (PI(3)K) signalling, Wnt/b-catenin signalling, histones, ubiquitin- mediated proteolysis, and splicing (Fig. 2).
Supplementary Data, Table 4
- globally significant, frequency >= 1% for glioblastoma multiforme (GBM): 46
Let’s use g:Profiler to obtain enrichment results
First set the parameters and filter gene sets to be analysed:

Note that input genes are ordered according to p-value so the Ordered Query option is appropriate.
Then paste the GBM gene list and press g:Profile to start the analysis.
Scroll down to see significantly enriched pathways and processes. Scroll right to see gene annotations of GO processes (colored) and Reactome pathways (black; scroll further down).

Browse results:
- Click on numbers in the column n. of common genes to find genes that are part of a given process.
- Click graph icon to see how processes are related to each other.
- Check checkbox No electronic GO annotations and run query again. Now you see the enrichment analysis that uses only high-confidence gene annotations.
- Uncheck checkbox Hierarchical sorting to reveal ranking of results by corrected p-value.
g:Convert - gene ID conversion:
- Paste gene list into the Query box.
- In the Target Database list, select the desired type of gene/protein identifiers, for example UNIPROTSWISSPROT. Click Convert IDs to continue.
For Enrichment Map construction:
- Set Output Type to Generic Enrichment Map (TAB).
- Click on g:Profile to run analysis again.
- Right-click on Download data in Generic Enrichment Map (GEM) format to save the file.
- Browse the downloaded file in a text editor. Note lists of genes in the leftmost column. These genes are part of the input list and responsible for the given pathway enrichment.
- At the bottom of Advanced Options, find Download g:Profiler data as GMT and right-click the link name to save the zip file with gene-set annotations.
- In the zip file, you will need the file hsapiens.pathways.NAME.gmt.
Building an Enrichment Map visualization in Cytoscape
- From the main menu, select Apps>Enrichment Map>Create Enrichment Map. If you don’t have this app, install version 2.2.1 using Cytoscape App Manager or Cytoscape App Store.
- First click on (+) to add a new analysis. Load all the files:
- GMT file: has gene-set definitions
- Enrichment of data-set: has enrichment statistics for GBM gene list
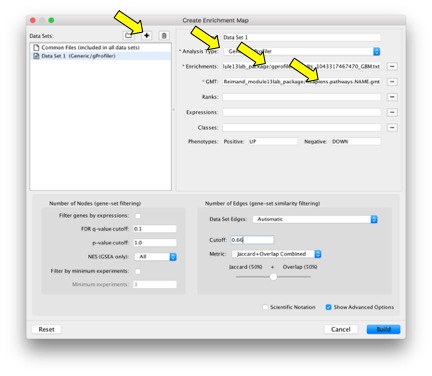
- Set analysis parameters under Advanced Options.
a. FDR-corrected P-values are filtered in g:Profiler and no further filtering is needed in Enrichment map.
b. Jaccard coefficient + overlap combined defines the stringency edges between gene sets and the granularity of the map. Use this parameter to create denser or sparser networks. The value 0.66 provides relatively stringent similarity cutoffs and leads to sparser maps.
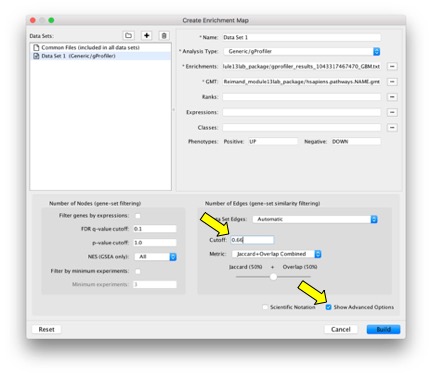
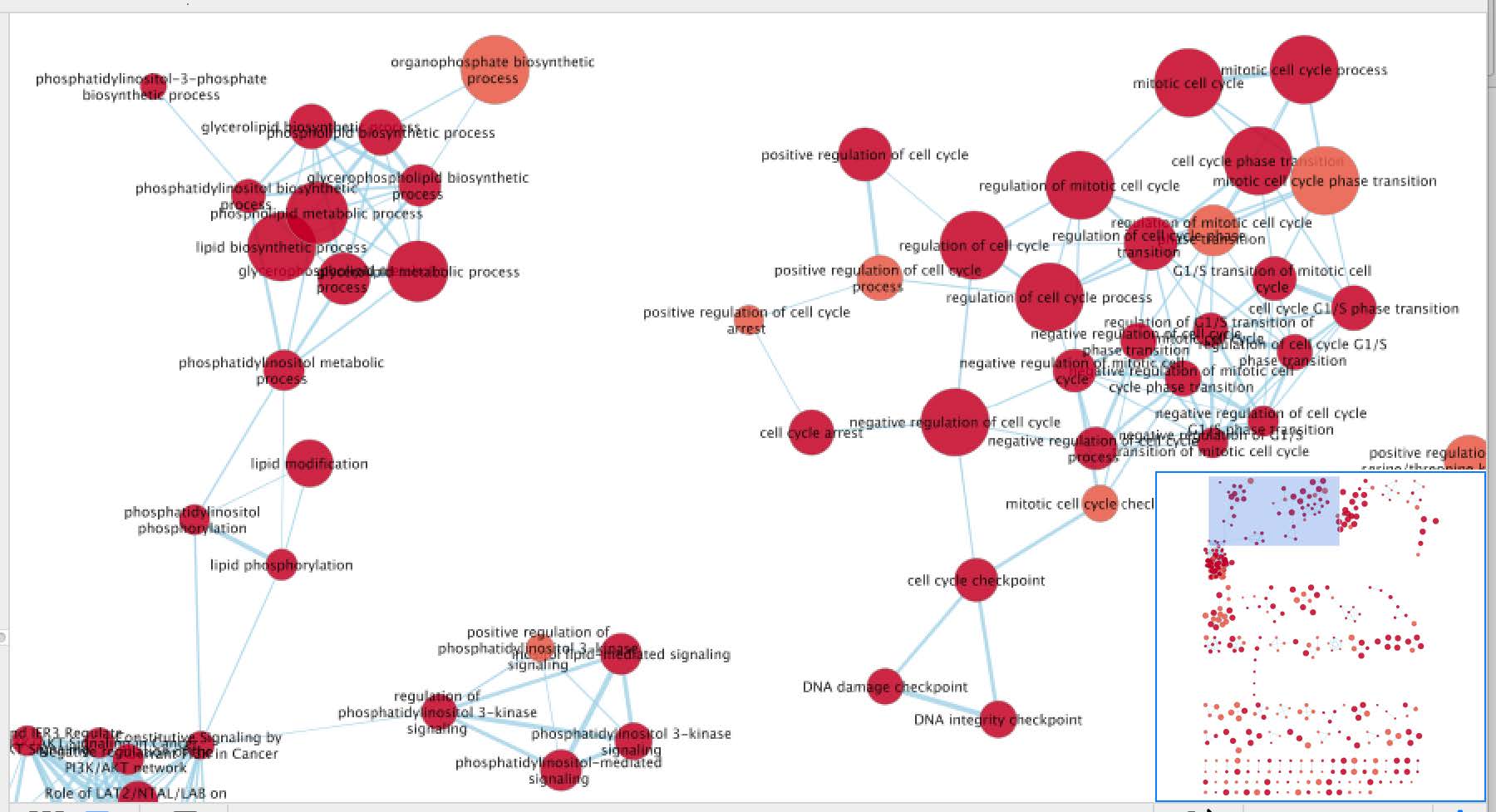
- Click ‘Build’. An enrichment map will be generated.
- The resulting map shows groups of enriched pathways. Pathways are connected by edges if they share a large fraction of related genes. That fraction is determined in point 3b above.